



















IMPORTANT SAFETY INFORMATION
Contraindication: EBGLYSS is contraindicated in patients with prior serious hypersensitivity to lebrikizumab-lbkz or any excipients of EBGLYSS.
Warnings and Precautions
Hypersensitivity
Hypersensitivity reactions, including angioedema and urticaria, have been reported with use of EBGLYSS. If a serious hypersensitivity reaction occurs, discontinue EBGLYSS and institute appropriate therapy.
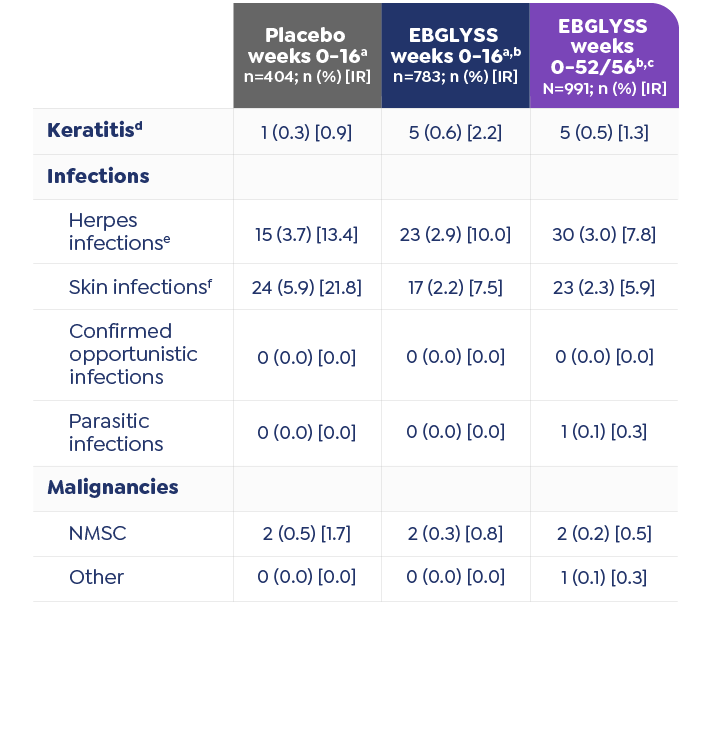
Conjunctivitis and Keratitis
Conjunctivitis and keratitis adverse reactions have been reported in clinical trials. Conjunctivitis and keratitis occurred more frequently in atopic dermatitis subjects who received EBGLYSS compared to those who received placebo. Conjunctivitis was the most frequently reported eye disorder. Most subjects with conjunctivitis or keratitis recovered during the treatment period. Advise patients to report new onset or worsening eye symptoms to their healthcare provider.
Parasitic (Helminth) Infections
Patients with known helminth infections were excluded from participation in clinical studies. It is unknown if EBGLYSS will influence the immune response against helminth infections by inhibiting IL-13 signaling. Treat patients with pre-existing helminth infections before initiating treatment with EBGLYSS. If patients become infected while receiving EBGLYSS and do not respond to antihelminth treatment, discontinue treatment with EBGLYSS until the infection resolves.
Vaccinations
EBGLYSS may alter a patient’s immunity and increase the risk of infection following administration of live vaccines. Prior to therapy with EBGLYSS, complete all age-appropriate vaccinations according to current immunization guidelines. Avoid use of live vaccines immediately prior to or during treatment with EBGLYSS. No data are available on the response to live vaccines.
Adverse Reactions
The most common (≥1%) adverse reactions are conjunctivitis, injection site reactions, and herpes zoster.
EBGLYSS is available as a 250mg/2mL subcutaneous injection prefilled pen or prefilled syringe.
Please click to access Prescribing Information and Patient Information.
Please see Instructions for Use included with the device.
LK HCP ISI AD APP
